[toc]
我从ubuntu官网下载了ubuntu20.04版本的系统iso文件,我是想将这个iso镜像按照自己的需求进行配置,最后形成一个可以随时将这个文件移植到任何电脑的一个系统。
step1:vbox安装iso文件
这部分教程参考:https://www.cnblogs.com/rocedu/p/6012545.html
这是很详细的教程照着做就好了。有以下几点注意一下:
设置共享剪切板的拖放
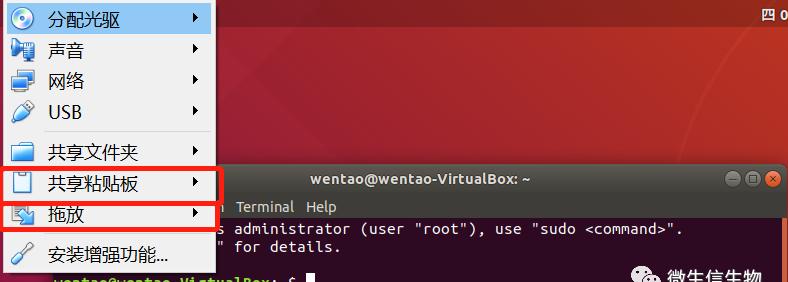 -作为分享的虚拟机,我的密码是:
-作为分享的虚拟机,我的密码是:
wentao
注意:上来第一个问题就是虚拟机屏幕显示大小问题:当开始只能显示一个很小的界面:先不用管,注意后续安装增强工鞥那可以解决问题。
step1:设置root密码sudo passwd#在提示下两次输入密码#后完成设置镜像设置ubuntu18镜像#备份sudo cp /etc/apt/sources.list /etc/apt/sources.list.bak#打开文件替换源sudo gedit /etc/apt/sources.list#在/etc/apt/sources.list文件中将全部文件替换成下面内##清华源deb https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic main restricted universe multiversedeb-src https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic main restricted universe multiversedeb https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic-updates main restricted universe multiversedeb-src https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic-updates main restricted universe multiversedeb https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic-backports main restricted universe multiversedeb-src https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic-backports main restricted universe multiversedeb https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic-security main restricted universe multiversedeb-src https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic-security main restricted universe multiversedeb https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic-proposed main restricted universe multiversedeb-src https://mirrors.tuna.tsinghua.edu.cn/ubuntu/ bionic-proposed main restricted universe multiverse##更新sudo apt-get updatesudo apt-get upgradeubuntu20镜像 (更新)#备份sudo cp /etc/apt/sources.list /etc/apt/sources.list.bak#打开文件替换源sudo gedit /etc/apt/sources.list#在/etc/apt/sources.list文件中将全部文件替换成下面内deb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal main restricteddeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal-updates main restricteddeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal universedeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal-updates universedeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal multiversedeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal-updates multiversedeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal-backports main restricted universe multiversedeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal-security main restricteddeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal-security universedeb http://mirrors.tuna.tsinghua.edu.cn/ubuntu/ focal-security multiverse##更新sudo apt-get updatesudo apt-get upgradestep2:conda安装
首先安装conda:
https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/我下载了一个最新版的linux版本到Shared_Folder中的src文件夹中。

这里的conda文件名称需要更换为自己的操作。
#2、提权chmod x Miniconda3-4.5.11-Linux-x86_64.sh#3、运行安装./Miniconda3-4.5.11-Linux-x86_64.sh #按照提示输入:enter-yes-yes#安装完成重启terminal生效
补充一批镜像
conda config –add channels conda-forgeconda config –add channels defaultsconda config –add channels rconda config –add channels biocondaconda config –add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/#最后一个是换国内的镜像#查看已经添加的channelsconda config –get channels安装谷歌浏览器
下载 google-chrome-stable_current_amd64.deb ,执行如下代码
then run :
sudo dpkg -i google-chrome-stable_current_amd64.deb安装conda环境
这里我们这个镜像用于多组学数据处理,所以我需要按照不同的组学安装不同的虚拟环境
首先是 扩增子数据分析
Qiime为代表的python2镜像及其工具
安装Qiime1
使用conda虚拟环境安装
conda create -n qiime1 python=2.7 qiime matplotlib=1.4.3 mock nose -c bioconda
提示错误,找不到qiime了,这个镜像难道不可以安装了吗?
在今年上半年我还安装过一次(发现是conda镜像没有很好的指定导致的-错误信息已在前面conda设置下载进项除做了更改)
#查看当前环境 conda info –envs#激活环境source activate qiime1#查看命令qiime –help#关闭环境conda deactivate安装qiime2
Qiime2为代表的python3镜像及其工具
断了好几次之后终于安装成功了
也是用虚拟环境https://docs.qiime2.org/2019.4/install/native/#install-qiime-2-within-a-conda-environment
wget https://data.qiime2.org/distro/core/qiime2-2019.10-py36-linux-conda.ymlconda env create -n qiime2-2019.10 –file qiime2-2019.10-py36-linux-conda.yml#激活环境source activate qiime2-2019.4#关闭环境conda deactivatevsearch usearch32位conda install vsearchR和rstudio (R语言已跟新到4.0)
R4.0 安装方法 ubuntu https://rtask.thinkr.fr/installation-of-r-4-0-on-ubuntu-20-04-lts-and-tips-for-spatial-packages/
sudo apt install apt-transport-https software-properties-common#sudo apt-key adv –keyserver keyserver.ubuntu.com –recv-keys E298A3A825C0D65DFD57CBB651716619E084DAB9#sudo add-apt-repository ‘deb https://cloud.r-project.org/bin/linux/ubuntu bionic-cran35/’sudo add-apt-repository ‘deb https://cloud.r-project.org/bin/linux/ubuntu focal-cran40/’sudo apt install libclang-dev# 密匙会经常失败,使用下面方式配制#sudo apt-key adv –keyserver keyserver.ubuntu.com –recv-keys E298A3A825C0D65DFD57CBB651716619E084DAB9sudo apt updategpg –keyserver hkp://keyserver.ubuntu.com:80 –recv-keys E298A3A825C0D65DFD57CBB651716619E084DAB9gpg -a –export E298A3A825C0D65DFD57CBB651716619E084DAB9 | sudo apt-key add -sudo apt install r-base r-base-core r-recommended r-base-dev
这时候R语言成功安装到环境中了
但是这次我安装的是1.2版本
# Ubuntu 18.04 / Linux Mint 19sudo apt -y install wgetwget https://download1.rstudio.org/desktop/bionic/amd64/rstudio-1.2.5019-amd64.debsudo dpkg -i rstudio-1.2.1578-amd64.debinstall rstudio serve
略
sudo rstudio-server startsudo rstudio-server status
运行 rstudio-server
http://127.0.0.1:8787/安装R包
在安装R包之前先安装一下几个依赖:
sudo apt-get install libcurl4-openssl-devsudo apt-get -y build-dep libcurl4-gnutls-devsudo apt-get install libcurl4-gnutls-devsudo apt-get install libxml2-devsudo apt-get install libcurl4-openssl-devsudo apt-get install libudunits2-devsudo apt-get install libgdal-devsudo apt-get install gfortransudo apt-get install build-essentialsudo apt-get install libxt-devsudo apt-get install libcurl4-openssl-devsudo apt-get install libxml 2.6-devsudo apt-get install libssl-devsudo apt-get install libxml2-devsudo apt-get install libfontconfig1-devsudo apt-get install libcairo2-devsudo apt-get install libxt-devsudo apt-get install libopenmpi-dev如何处理错误
How to Fix ‘E: Could not get lock /var/lib/dpkg/lock’ Error in Ubuntu Linux
N: Be aware that removing the lock file is not a solution and may break your system. E: Unable to acquire the dpkg frontend lock (/var/lib/dpkg/lock-frontend), is another process using it?
处理方法
ps aux | grep -i aptsudo kill 740install.packages(“devtools”, dependencies = T)library(devtools)
各种依赖需要解决,所以还是挺麻烦的;这里献上我的代码(基本布置的环境,后期根据需求在安装一些其他的包):
install(“phyloseq”)install(“microbiome”)install(“microbiomeSeq”)umerijaz/microbiomeSeq
现在一个基于扩增子分析的vdi虚拟镜像就做好了。下一节,我们将带大家继续升级这个镜像文件;
安装搜狗输入法
出现乱码:在terminal中输入:
killall fcitx
然后重启fcitx,后可用
问题集锦/第二次配置错误1:我发现共享文件夹无法安装
首先共享文件夹无法安装增强工具:
也不是为了熟悉这个过程,实在是第一个系统崩溃了,所以我有进行第二次的重新尝试,同样发现增强功能还是安装不上,这可能是普遍问题??
#报错信息如下This system is currently not set up to build kernel modules.Please install the gcc make perl packages from your distribution.VirtualBox Guest Additions: Running kernel modules will not be replaced untilthe system is restartedVirtualBox Guest Additions: Starting.VirtualBox Guest Additions: Building the modules for kernel 4.18.0-15-generic.This system is currently not set up to build kernel modules.Please install the gcc make perl packages from your distribution.Press Return to close this window…
如下解决:
# 手动安装gcc make perl模块sudo apt install perlsudo apt install makesudo apt install gcc#提示这个模块已有
从起电脑,我将虚拟盘片弹出,然后重新run,因为不能挂在两次,在重新安装增强工具—得以成功
错误2:无法安装
一下模块需要安装,但是却无法安装,这是由于镜像没有设置好导致的,所以之前的那个清华的镜像一定要弄好。要不然啥软件都不能安装。
# 手动安装gcc make perl模块sudo apt install perl错误3: 刚安装的ubuntu虚拟机窗口界面是很小的?无法修改?
但是在安装完成增强功能后就适应了电脑屏幕大小
错误4:下一个问题:共享文件夹带锁
我想这个问题之前没有遇到,在网上找了一圈,还是没有发现答案,就去外面找了,果然马上就找到方案了:
https://stackoverflow.com/questions/26740113/virtualbox-shared-folder-permissions
sudo adduser $USER vboxsf
重启—-得以解决,注意需要重启
经常去外面逛逛还是好的。
问题5:R和Rstudio之间关联问题
sudo apt install apt-transport-https software-properties-commonsudo apt-key adv –keyserver keyserver.ubuntu.com –recv-keys E298A3A825C0D65DFD57CBB651716619E084DAB9sudo add-apt-repository ‘deb https://cloud.r-project.org/bin/linux/ubuntu bionic-cran35/’#跟新sudo apt update#使用conda安装R解决依赖问题conda install R
这时候R语言成功安装到环境中了
rstudio和rstudio_sever的安装的安装-安装失败-换用conda装
conda install rstudio -c bioconda
尚未关联错误:
unable to find installation of R on the system (which R did return valid output);unable to lacate R binary by scanning standard lacations
这时候Rstudio也成功安装到环境中了,此时在terminal中可以启动Rstudio,但是在系统中却找不到rstudio的快捷方式。
失败安装过程
rstudio无法和R关联,所以我选择同时使用conda安装两者。首先安装 gdebi
rstudio_sever安装
我先不安装,rstudi官网安装指导:https://rstudio.com/products/rstudio/download-server/debian-ubuntu/
sudo apt-get install gdebi-corewget https://download2.rstudio.org/server/trusty/amd64/rstudio-server-1.2.5001-amd64.debsudo gdebi rstudio-server-1.2.5001-amd64.deb#查看ipifconfig -a
其这次我安装studio
wget https://download1.rstudio.org/rstudio-xenial-1.1.447-amd64.debsudo gdebi rstudio-xenial-1.1.447-amd64.deb
以上过程够尝试过,发现还是没办法解决问题
#卸载rstudiosudo apt-get –purge remove
使用apt错误,但是重新启动terminal就好了
E: Could not get lock /var/lib/dpkg/lock-frontend – open (11: Resource temporarily unavailable)E: Unable to acquire the dpkg frontend lock (/var/lib/dpkg/lock-frontend), is another process using it?R包安装错误 microbiomeSeq
这个问题到现在也不知道是什么原因:
我在github上提问了一下:https://github.com/umerijaz/microbiomeSeq/issues/41
Error: package or namespace load failed for ‘microbiomeSeq’: object ‘bringToTop’ is not exported by ‘namespace:grDevices’Error when install rstudio(Reading database … 170206 files and directories currently installed.)Preparing to unpack rstudio-1.2.5019-amd64.deb …Unpacking rstudio (1.2.5019) over (1.2.5019) …dpkg: dependency problems prevent configuration of rstudio: rstudio depends on libclang-dev; however: Package libclang-dev is not installed.dpkg: error processing package rstudio (–install): dependency problems – leaving unconfiguredProcessing triggers for gnome-menus (3.13.3-11ubuntu1.1) …Processing triggers for desktop-file-utils (0.23-1ubuntu3.18.04.2) …Processing triggers for mime-support (3.60ubuntu1) …Processing triggers for hicolor-icon-theme (0.17-2) …Processing triggers for shared-mime-info (1.9-2) …Errors were encountered while processing: rstudio
but when we install:
sudo apt-get install libclang-dev
another error occour :
Reading package lists… DoneBuilding dependency treeReading state information… DoneYou might want to run ‘apt –fix-broken install’ to correct these.The following packages have unmet dependencies: libclang-dev : Depends: libclang-6.0-dev (>= 6.0~) but it is not going to be installedE: Unmet dependencies. Try ‘apt –fix-broken install’ with no packages (or specify a solution).
so i must install :
sudo apt –fix-broken install
then, libclang-dev and rstudio were installed successfully
Errorconda create -n qiime1 python=2.7 qiime matplotlib=1.4.3 mock nose -c biocondaWARNING: A conda environment already exists at ‘/home/wentao/miniconda3/envs/qiime1’Remove existing environment (y/[n])? nRmpi R packageconfigure: error: “Cannot find mpi.h header file” ERROR: configuration failed for package ‘Rmpi’sudo apt-get install libopenmpi-deverror Cairo R packageconfigure: error: Cannot find cairo.h! Please install cairo (http://www.cairographics.org/) and/or ssudo apt-get install libcairo2-devsudo apt-get install libxt-dev#instal R package menegerinstall.packages(“BiocManager”)library(“BiocManager”)install.packages(“devtools”, dependencies = T)library(devtools)install_github(“umerijaz/microbiomeSeq”)# install big R packagesinstall(“phyloseq”)library(phyloseq)BiocManager::install(“microbiome”)library(“microbiome”)BiocManager::install(“microbiomeSeq”)library(“microbiomeSeq”)BiocManager::install(“DESeq2”)library(“DESeq2”)BiocManager::install(“impute”)BiocManager::install(“preprocessCore”)BiocManager::install(“GO.db”)BiocManager::install(“adespatial”)BiocManager::install(“units”)BiocManager::install(“spdep”)BiocManager::install(“adegenet”)BiocManager::install(“adephylo”) .libPaths()#得到所有包的文件路径 #在terminl中输入,讲这些路径全部可用 sudo chmod -R 777 “/home/wentao/R/x86_64-pc-linux-gnu-library/3.6” sudo chmod -R 777 “/usr/local/lib/R/site-library” sudo chmod -R 777 “/usr/lib/R/site-library” sudo chmod -R 777 “/usr/lib/R/library” devtools::install_github(“xia-lab/MetaboAnalystR”) library(“MetaboAnalystR”) metr_pkgs <- c(“Rserve”, “ellipse”, “scatterplot3d”, “Cairo”, “randomForest”, “caTools”, “e1071”, “som”, “impute”, “pcaMethods”, “RJSONIO”, “ROCR”, “globaltest”, “GlobalAncova”, “Rgraphviz”, “preprocessCore”, “genefilter”, “pheatmap”, “SSPA”, “sva”, “Rcpp”, “pROC”, “data.table”, “limma”, “car”, “fitdistrplus”, “lars”, “Hmisc”, “magrittr”, “methods”, “xtable”, “pls”, “caret”, “lattice”, “igraph”, “gplots”, “KEGGgraph”, “reshape”, “RColorBrewer”, “tibble”, “siggenes”, “plotly”) list_installed <- installed.packages() new_pkgs <- subset(metr_pkgs, !(metr_pkgs %in% list_installed[, “Package”])) if(length(new_pkgs)!=0){ # source(“https://bioconductor.org/biocLite.R”) BiocManager::install(new_pkgs, dependencies = TRUE, ask = FALSE) print(c(new_pkgs, ” packages added…”)) } BiocManager::install(“systemfonts”)install.packages(“Rmpi”)install.packages(“Cairo”) BiocManager::install(“systemfonts”) BiocManager::install(“xcms”) BiocManager::install(“CAMERA”) BiocManager::install(“fgsea”) BiocManager::install(“MSnbase”) BiocManager::install(“systemfonts”)# fontconfig Rmpi gdtools freetypeharfbuzz rgl vdiffr